Abstract
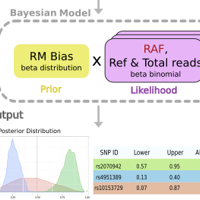
Allele-specific measurements of transcription factor binding from ChIP-seq data are key to dissecting the allelic effects of non-coding variants and their contribution to phenotypic diversity. However, most methods of detecting an allelic imbalance assume diploid genomes. This assumption severely limits their applicability to cancer samples with frequent DNA copy-number changes. Here we present a Bayesian statistical approach called BaalChIP to correct for the effect of background allele frequency on the observed ChIP-seq read counts. BaalChIP allows the joint analysis of multiple ChIP-seq samples across a single variant and outperforms competing approaches in simulations. Using 548 ENCODE ChIP-seq and six targeted FAIRE-seq samples, we show that BaalChIP effectively corrects allele-specific analysis for copy-number variation and increases the power to detect putative cis-acting regulatory variants in cancer genomes.