Abstract
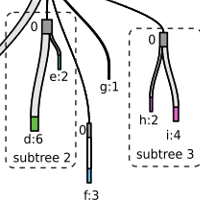
Cancer has long been understood as a somatic evolutionary process, but many details of tumor progression remain elusive. Here, we present BitPhylogeny, a probabilistic framework to reconstruct intra-tumor evolutionary pathways. Using a full Bayesian approach, we jointly estimate the number and composition of clones in the sample as well as the most likely tree connecting them. We validate our approach in the controlled setting of a simulation study and compare it against several competing methods. In two case studies, we demonstrate how BitPhylogeny reconstructs tumor phylogenies from methylation patterns in colon cancer and from single-cell exomes in myeloproliferative neoplasm.