Abstract
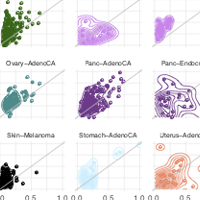
We present SVclone, a computational method for inferring the cancer cell fraction of structural variant breakpoints from whole-genome sequencing data. We validate our approach using simulated and real tumour samples, and demonstrate its utility on 2,778 whole-genome sequenced tumours. We find a subset of liver, breast and ovarian cancer cases with decreased overall survival that have subclonally enriched copy-number neutral rearrangements, an observation that could not be discovered with currently available methods.