Abstract
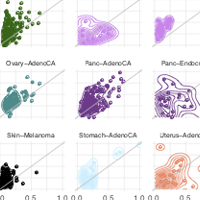
We present SVclone, a computational method for inferring the cancer cell fraction of structural variant (SV) breakpoints from whole-genome sequencing data. SVclone accurately determines the variant allele frequencies of both SV breakends, then simultaneously estimates the cancer cell fraction and SV copy number. We assess performance using in silico mixtures of real samples, at known proportions, created from two clonal metastases from the same patient. We find that SVclone’s performance is comparable to single nucleotide variant-based methods, despite having an order of magnitude fewer data points. As part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) consortium, which aggregated whole genome sequencing data from 2,658 cancers across 38 tumour types, we apply SVclone to these data and reveal a subset of liver, ovarian and pancreatic cancers with decreased overall survival and subclonally enriched copy number neutral rearrangements, demonstrating that the clonality of balanced rearrangements can reveal clinically relevant observations.